Genetic Mutation Age Estimator
An R Shiny app based on the mutation dating method developed in the paper Dating Rare Mutations from Small Samples with Dense Marker Data.
This application will estimate the age of a genetic mutation, based on the genetic length of ancestral haplotypes common to individuals who share the mutation. Although originally developed for dating of rare mutations in human populations, this method can be applied to any species for which a genetic map is available.
In publications, please cite this website, and the following paper:
Citation: Luke C. Gandolfo, Melanie Bahlo and Terence P. Speed. Dating Rare Mutations from Small Samples with Dense Marker Data. Genetics August 1, 2014 vol. 197 no. 4 1315-1327; https://doi.org/10.1534/genetics.114.164616
Usage Information
Obtain shared ancestral segments regions
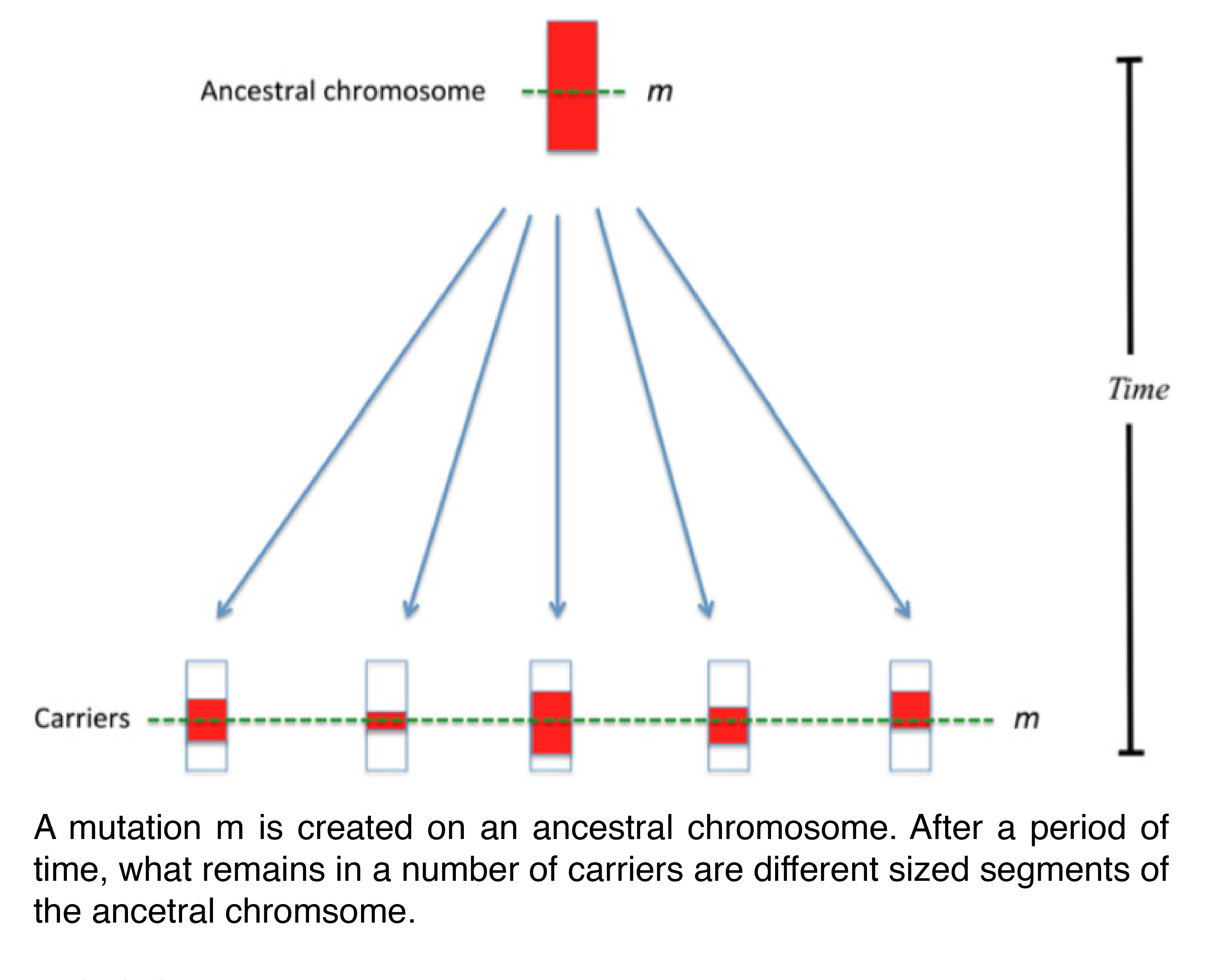
Firstly, use the following procedure to obtain the genetic length of ancestral segments in sampled individuals (see Figure 1):
- Line up your high density SNP data around the mutation locus.
- Highlight continuous haplotype sharing between two or more individuals as you move away from the mutation locus. This sharing defines the ancestral segments.
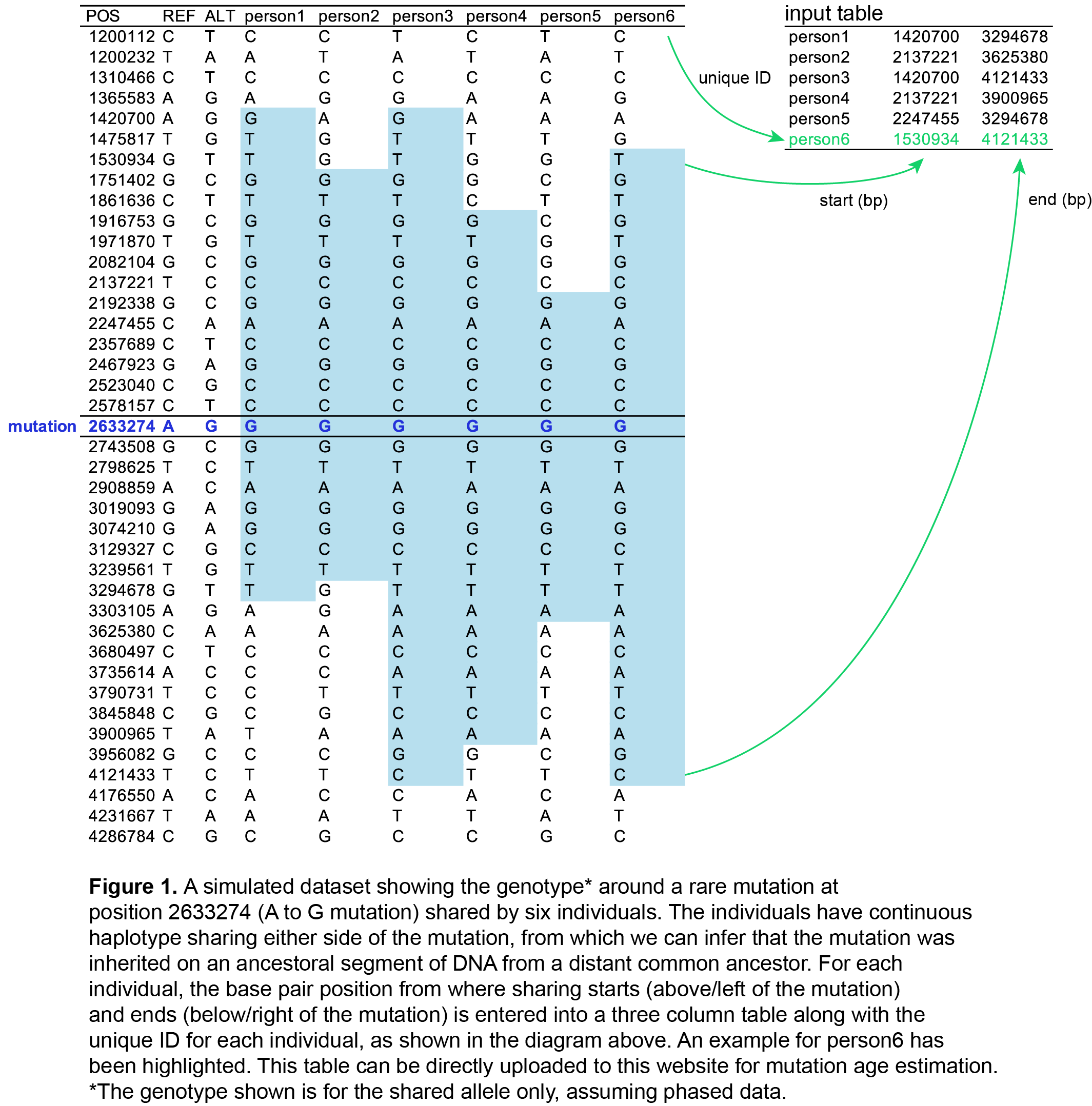
- Summarise the shared regions in a three column table (tab separated) with the following columns for each person:
- column 1: unique individual ID. No specific formatting required.
- column 2: the genomic position in base pairs of the start of the ancestral sharing for that individual (i.e. the first base position of the shaded region in Fig. 1).
- column 3: the genomic position in base pairs of the end of the ancestral sharing for that individual (i.e. the last base position of the shaded region in Fig. 1).
To assess haplotype sharing for individuals with a single copy of the mutation (e.g. with a dominant mutation), or for individuals with copies of two distinct mutations at the same gene locus (i.e. with a compound heterozygous mutation), the data needs to be phased to resolve the underlying ancestral haplotype(s). However, for individuals with two copies of the same recessive mutation, phasing is not required: ancestral segments are defined by continuous sharing of identical homozygous markers.
Default input options
Default options available for human data only, in hg19 reference genome.
Upload file of shared ancestoral regions (hg19): upload a tab-seperated file (with no header) table of the shared ancestral regions for each individual (see Figure 1 for example).
Chromosome: select the chromosome on which the mutation is located.
Genomic position of mutation (bp): enter the position, in bp, of mutation. If the mutation spans more than one bp, use the start position.
Advanced input options
Input text (all species) or Upload file (all species) can be used if the ancestral arm lengths have already been calculated and converted to centiMorgans (cM). These options can be used for both human and non-human data, as long as a genomic map (with postions recorded in cM) is available for the species.
Ancestral lengths (cM): if you have already determined the ancestral arm lengths in cM, ancestral lengths (both left and right arms) can be directly uploaded as a csv file, or entered directly as text:
- For the list of left arm lengths, input the data as follows: left arm length for individual 1, ... , left arm length for individual n.
- For the list of right arm lengths, input the data as follows: right arm length for individual 1, ... , right arm length for individual n.
Note that the components of each list must correspond to the same individuals, as shown above. Lengths must be in units of centiMorgans (cM). CSV files must not contain any column or row names.
To obtain ancestral lengths: using a genetic map, record the genetic length of the segments. Specifically: record the segment “arm” lengths for each individual, i.e. the left arm length is the genetic distance between the mutation and the outermost marker heading in the direction of decreasing map position and the right arm length is the distance between the mutation and the outermost marker heading in the direction of increasing map position. Record the lengths in units of cM.
Confidence interval: a value between 0 and 1. Defaults to 0.95 (i.e. 95% confidence interval).
Chance sharing: select TRUE to adjust for chance sharing, otherwise FALSE. If chance sharing is true, please specify the following parameters (for the chromosome on which the mutation is located):
- Median allele frequency: a number between 0 and 1.
- Markers on chromosome: the number of markers on the chromosome.
- Length of chromosome: the genetic length (in cM) of the chromosome.
Dating mutations results:
Summary of results:
Example results:
Click 'Submit Example' to see example results
Summary of results:
Genealogy
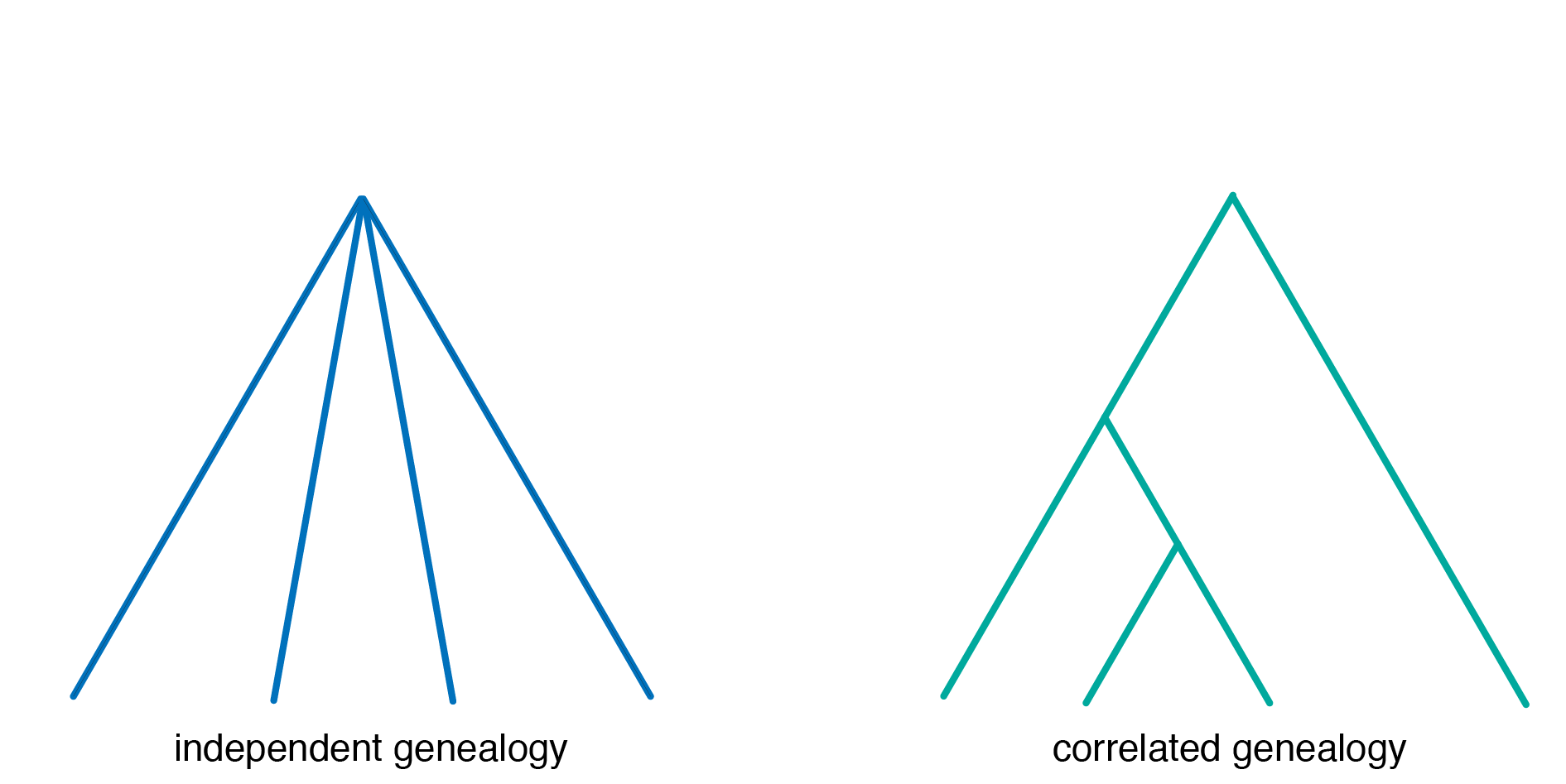
This method returns the age in generations for both independent and correlated genealogies.
- independent: sampled individuals have independent recombinational histories since their most recent common ancestor (MRCA).
- correlated: subsets of the sampled individuals have common ancestry earlier than the MRCA for the entire sample, i.e. a “tree-like” genealogy.
However, please note that genealogies are rarely independent, and unless there is information to indicate otherwise, genealogies should be assumed to be correlated.
For further information regarding independent and correlated genealogy age estimates, please refer to:
Luke C. Gandolfo, Melanie Bahlo and Terence P. Speed. Dating Rare Mutations from Small Samples with Dense Marker Data. Genetics August 1, 2014 vol. 197 no. 4 1315-1327
Citations
Full citation list is available here: Google scholar citations
This publication has been cited by 39 papers (as of Jan 2022), including the following papers:
- Park, Y.H., Remmers, E.F., Lee, W. et al. Ancient familial Mediterranean fever mutations in human pyrin and resistance to Yersinia pestis . Nat Immunol . 2020;21:857–67. https://doi.org/10.1038/s41590-020-0705-6
- Florian, R.T., Kraft, F., Leitão, E. et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3 . Nat Commun . 2019;10:4919. https://doi.org/10.1038/s41467-019-12763-9
- Alistair T Pagnamenta, Rauan Kaiyrzhanov, Yaqun Zou, et al. An ancestral 10-bp repeat expansion in VWA1 causes recessive hereditary motor neuropathy . Brain . 2021;144(2):584-600 https://doi.org/10.1093/brain/awaa420