Human CNS gene expression & RNA editing browser
Welcome to the CNS browser. Created by Brendan Ansell, Bahlo Laboratory, Walter + Eliza Hall Institute. In this browser you can investigate gene expression levels in single neuronal nuclei assayed in the studies below, as well as RNA editing events in those nuclei.
Please use the tabs to search for your gene or editing site of interest
Abstracts of represented publications
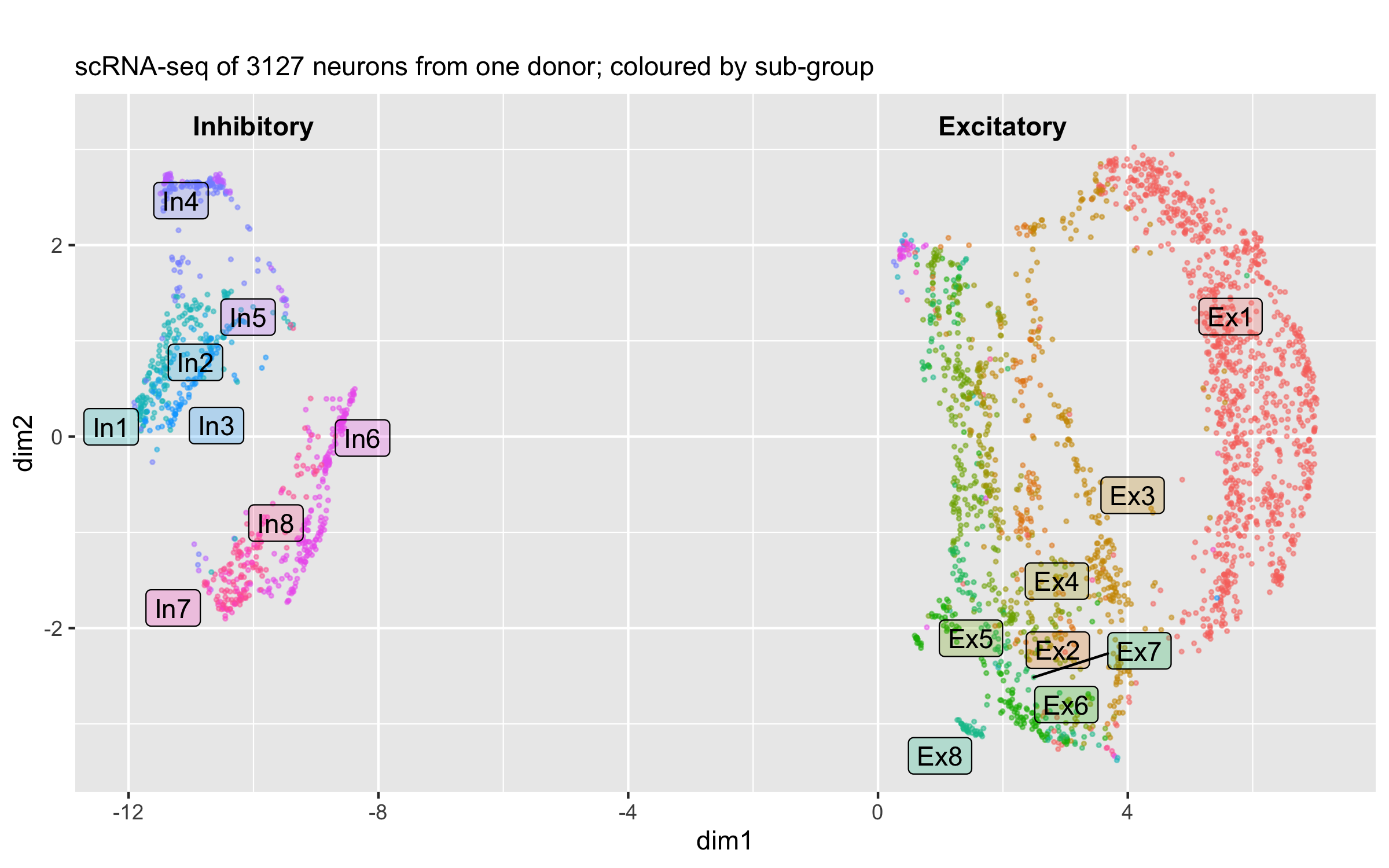
Lake et al Science 2016
The human brain has enormously complex cellular diversity and connectivities fundamental to our neural functions, yet difficulties in interrogating individual neurons has impeded understanding of the underlying transcriptional landscape. We developed a scalable approach to sequence and quantify RNA molecules in isolated neuronal nuclei from a postmortem brain, generating 3227 sets of single-neuron data from six distinct regions of the cerebral cortex. Using an iterative clustering and classification approach, we identified 16 neuronal subtypes that were further annotated on the basis of known markers and cortical cytoarchitecture. These data demonstrate a robust and scalable method for identifying and categorizing single nuclear transcriptomes, revealing shared genes sufficient to distinguish previously unknown and orthologous neuronal subtypes as well as regional identity and transcriptomic heterogeneity within the human brain.
Darmanis et al PNAS 2015
The human brain is a tissue of vast complexity in terms of the cell types it comprises. Conventional approaches to classifying cell types in the human brain at single cell resolution have been limited to exploring relatively few markers and therefore have provided a limited molecular characterization of any given cell type. We used single cell RNA sequencing on 466 cells to capture the cellular complexity of the adult and fetal human brain at a whole transcriptome level. Healthy adult temporal lobe tissue was obtained during surgical procedures where otherwise normal tissue was removed to gain access to deeper hippocampal pathology in patients with medical refractory seizures. We were able to classify individual cells into all of the major neuronal, glial, and vascular cell types in the brain. We were able to divide neurons into individual communities and show that these communities preserve the categorization of interneuron subtypes that is typically observed with the use of classic interneuron markers. We then used single cell RNA sequencing on fetal human cortical neurons to identify genes that are differentially expressed between fetal and adult neurons and those genes that display an expression gradient that reflects the transition between replicating and quiescent fetal neuronal populations. Finally, we observed the expression of major histocompatibility complex type I genes in a subset of adult neurons, but not fetal neurons. The work presented here demonstrates the applicability of single cell RNA sequencing on the study of the adult human brain and constitutes a first step toward a comprehensive cellular atlas of the human brain.
Welcome to the CNS browser for healthy brain single-neuron RNA editing. Here you can explore RNA editing sites quantified in transcriptomes from single neuronal nuclei published by Lake et al, Science 2016.
References for the datasets used in this server are provided at the bottom of the page.
Enter your editing site of interest (Hg38 coordinates) in the box below, or scroll down to investigate editing across entire genes.
Edited allele proportion per cell
Grouped by neuronal subgroup
Grouped by cortical region
Scroll over points to reveal editing site details. Click and drag to zoom in to a region of the plot. Clicking on a point will update the editing site table above.
Reference datasets and publications
Darmanis et al, PNAS 2015Breen et al Nature Neuroscience 2019
Tran et al Nature Neuroscience 2019
Picardi et al Nucleic Acids Research 2017
Landrum et al Nucleic Acids Research 2018